How virus detectives trace the origins of an outbreak – and why it's so tricky
Marilyn J. Roossinck, Penn State
 |
| The prevention of future pandemics requires examining viral family trees. Stockcrafter/iStock via Getty Images Plus |
In order to predict and prevent future pandemics like COVID-19, researchers need to find the origin of the viruses that cause them. This is not a trivial task.
The origin of HIV was not clear until 20 years after it spread around the world. Scientists still don’t know the origin of Ebola, even though it has caused periodic epidemics since the 1970s.
As an expert in viral ecology, I am often asked how scientists trace the origins of a virus. In my work, I have found many new viruses and some well-known pathogens that infect wild plants without causing any disease. Plant, animal or human, the methods are largely the same. Tracking down the origins of a virus involves a combination of extensive fieldwork, thorough lab testing and quite a bit of luck.
Viruses jump from wild animal hosts to humans
Many viruses and other disease agents that infect people originate in animals. These diseases are zoonotic, meaning they are caused by animal viruses that jumped to people and adapted to spread through the human population.
It might be tempting to start the viral origin search by testing sick animals at the site of the first known human infection, but wild hosts often don’t show any symptoms. Viruses and their hosts adapt to each other over time, so viruses often don’t cause obvious disease symptoms until they’ve jumped to a new host species. Researchers can’t just look for sick animals.
Another problem is that people and their food animals aren’t stationary. The place where researchers find the first infected person is not necessarily close to the place where the virus first emerged.

In the case of COVID-19, bats were an obvious first place to look. They’re known hosts for many coronaviruses and are the probable source of other zoonotic diseases like SARS and MERS.
For SARS-CoV-2, the virus that causes COVID-19, the nearest relative scientists have found so far is BatCoV RaTG13. This virus is part of a collection of bat coronaviruses discovered in 2011 and 2012 by virologists from the Wuhan Virology Institute.
The virologists were looking for SARS-related coronaviruses in bats after the SARS-CoV-1 pandemic in 2003. They collected fecal samples and throat swabs from bats at a site in Yunnan Province about 932 miles (1,500 kilometers) from the institute’s lab in Wuhan, where they brought samples back for further study.
To test whether the bat coronaviruses could spread into people, researchers infected monkey kidney cells and human tumor-derived cells with the Yunnan samples. They found that a number of the viruses from this collection could replicate in the human cells, meaning they could potentially be transmitted directly from bats to humans without an intermediate host. Bats and people don’t come into direct contact very often, however, so an intermediate host is still quite likely.
Finding the nearest relatives
The next step is to determine how closely related a suspected wildlife virus is to the one infecting humans. Scientists do this by figuring out the genetic sequence of the virus, which involves determining the order of the basic building blocks, or nucleotides, that make up the genome. The more nucleotides two genetic sequences share, the more closely related they are.
Genetic sequencing of bat coronavirus RaTG13 showed it to be over 96% identical to SARS-CoV-2. This level of similarity means that RaTG13 is a pretty close relative to SARS-CoV-2, confirming that SARS-CoV-2 probably originated in bats, but is still too distant to be a direct ancestor. There likely was another host that caught the virus from bats and passed it on to humans.

Because some of the earliest cases of COVID-19 were found in people associated with the wildlife market in Wuhan, there was speculation that a wild animal from this market was the intermediate host between bats and humans. However, researchers never found the coronavirus in animals from the market.
Likewise, when a related coronavirus was identified in pangolins confiscated in an anti-smuggling operation in southern China, many leaped to the conclusion that SARS-CoV-2 had jumped from bats to pangolins to humans. The pangolin virus was found to be only 91% identical to SARS-CoV-2, though, making it unlikely to be a direct ancestor of the human virus.
To pinpoint the origin of SARS-CoV-2, a lot more wild samples need to be collected. This is a difficult task – sampling bats is time-consuming and requires strict precautions against accidental infection. Since SARS-related coronaviruses are found in bats across Asia, including Thailand and Japan, it’s a very big haystack to search for a very small needle.
Creating a family tree for SARS-CoV-2
In order to sort out the puzzle of viral origins and movement, scientists not only have to find the missing pieces, but also figure out how they all fit together. This requires collecting viral samples from human infections and comparing those genetic sequences both to each other and to other animal-derived viruses.
To determine how these viral samples are related to each other, researchers use computer tools to construct the virus’s family tree, or phylogeny. Researchers compare the genetic sequences of each viral sample and construct relationships by aligning and ranking genetic similarities and differences.
The direct ancestor to the virus, sharing the greatest genetic similarity, could be thought of as its parent. Variants sharing that same parent sequence but with enough changes to make them distinct from each other are like siblings. In the case of SARS-CoV-2, the South African variant, B.1.351, and the U.K. variant, B.1.1.7, are siblings.
Building a family tree is complicated by the fact that different analysis parameters can give different results: The same set of genetic sequences can produce two very different family trees.
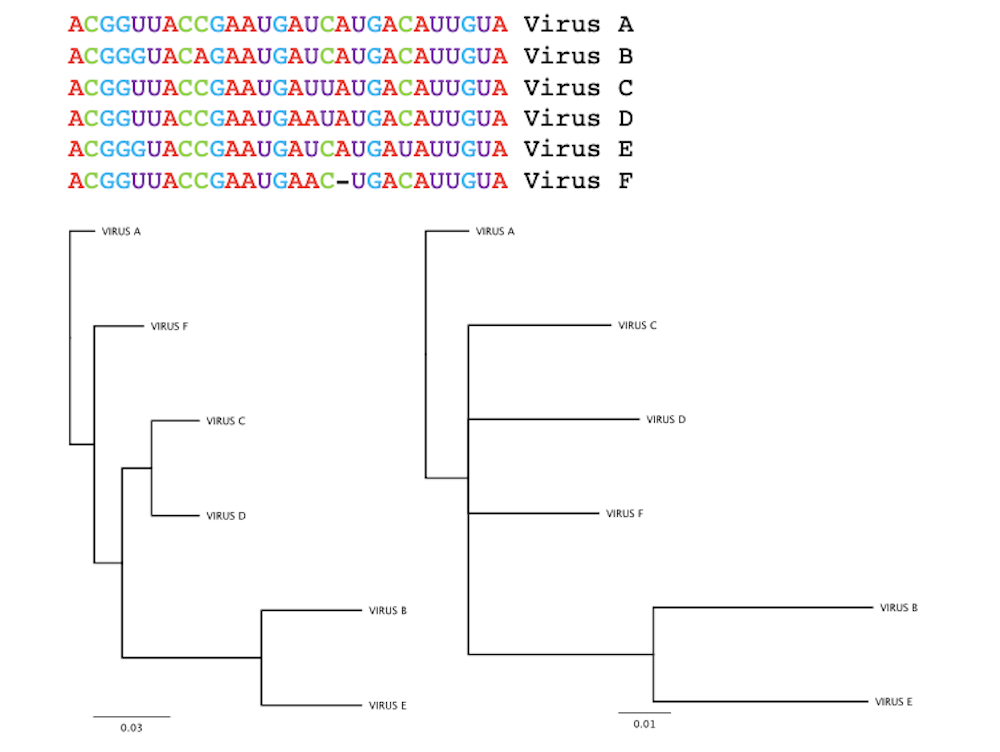
For SARS-CoV-2, phylogenetic analysis proves particularly difficult. Though tens of thousands of SARS-CoV-2 sequences are now available, they don’t differ from one another enough to form a clear picture of how they’re related to each other.
The current debate: Wild host or lab spillover?
Could SARS-CoV-2 have been released from a research lab? Although current evidence implies that this is not the case, 18 prominent virologists recently suggested that this question should be further investigated.
Although there has been speculation about SARS-CoV-2 being engineered in a lab, this possibility seems highly unlikely. When comparing the genetic sequence of wild RaTG13 with SARS-CoV-2, differences are randomly spread across the genome. In an engineered virus, there would be clear blocks of changes that represent introduced sequences from a different viral source.
There is one unique sequence in the SARS-CoV-2 genome that codes for a part of the spike protein that seems to play an important role in infecting people. Interestingly, a similar sequence is found in the MERS coronavirus that causes a disease similar to COVID-19.
Though it is not clear how SARS-CoV-2 acquired these sequences, viral evolution suggests they arose from natural processes. Viruses accumulate changes either by genetic exchange with other viruses and their hosts, or by random mistakes during replication.
Viruses that gain a genetic change that gives them a reproductive advantage would typically continue to pass it on through replication. That MERS and SARS-CoV-2 share a similar sequence in this part of the genome suggests that it naturally evolved in both and spread because it helps them infect human cells.
Where to go from here?
Figuring out the origin of SARS-CoV-2 could give us clues to understand and predict future pandemics, but we may never know exactly where it came from. Regardless of how the SARS-CoV-2 jumped into humans, it’s here now, and it’s probably here to stay. Going forward, researchers need to continue monitoring its spread, and get as many people vaccinated as possible.![]()
Marilyn J. Roossinck, Professor of Plant Pathology and Environmental Microbiology, Penn State
This article is republished from The Conversation under a Creative Commons license. Read the original article.
[Get our best science, health and technology stories. Sign up for The Conversation’s science newsletter.]